
The CESCG Stem Cell Hub is a data warehouse for stem cell genomics files produced through the CIRM Genomics Initiative. It houses primary data files such as DNA sequencing reads in fastq format, as well as many other file types derived from read mapping and analysis of the primary data, and PDF and other document files describing protocols. A small but flexible system, tag storm, for associating metadata information with a file.
Any CIRM Genomics Initiative associated lab can submit data to the Stem Cell Hub. Once submitted, data is treated as prepublication data, with access only allowed to authorized users (data privacy is described in more detail in the Privacy section below). Contact us if you are a Genomics Initiative lab and would like to submit data.
An account is not needed to access any of the data available through the Stem Cell Hub public site.
An account is needed to access data stored on our private server, which is intended for CESCG contributing labs to access their prepublication data. If you are associated with a contributing lab, please contact us for an account to access your data.
Once data is submitted to the Stem Cell Hub, access is only allowed to members of that lab. If you are part of a CIRM Genomics Initiative lab and would like access to your data, please contact us.
Once notified by the lab, the data will be released to the public meaning that anyone can download and access the data, even without an account. While this will be true for nearly all data, there will still be some data files that will need an approved account to access them. If there are files that you are interested in, but don't have access to you can request access to them.
The primary method for finding your data is through the File Search page, which can be found through "Browse > Files" in the menu at the top of the page.
The "Files" page by default displays a list of all available files in the Stem Cell Hub. The list of files can be filtered using the metadata facets on the left-hand side of the page.
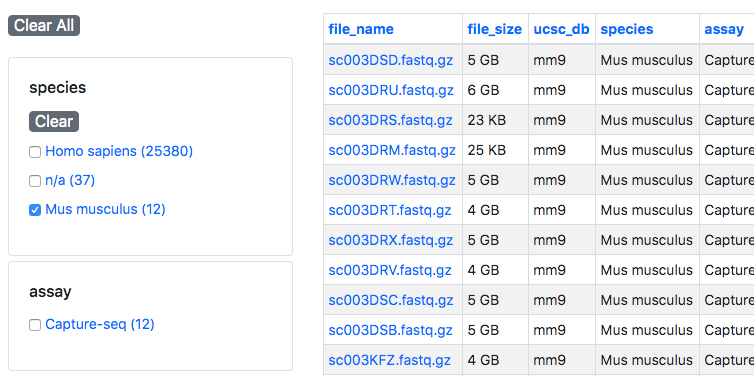
These metadata facets include the submitting lab, organism (e.g. human, mouse), and assay type (e.g. ATAC-seq, RRBS). By default, all of these facets are unselected and all available data in the Stem Cell hub is displayed. To show data that matches only a specific metadata value, check the box next to that value. The list of files will automatically be filtered to show only the matching files. Multiple facets can be combined to further filter the list of files.
The "Files" page also supports free-text search of metadata through the search box at the top:

Once you've filtered your files to the list of those you're interested in, you can download them.
Once you've filtered down the list of files to those that you are interested in, you can download them in one of two ways:
If you have only a few files to download, you can download them one-by-one. First, click the file name in the first column to be taken to the file details page.
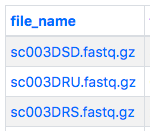
From there click the "download" link next to the name in the "accession" row to start the download. Files will be named after the Stem Cell Hub applied accession.
If you have many files to download, or a few large files that may take hours to download, you can use a variety of methods to download the files.
First, click the "Download All" link at the top of the page, from there you will be taken a page that lists the total number of files and their combined size as well as a few different download options:

Follow the instructions on that page to download the files. There are options for downloading your files using the command-line or web browser extensions. The URLs provided to you are valid for a week.
There are multiple ways to download and explore the metadata available in the scHub. These include the html/text/tsv/csv links on the datasets page:

And the Metadata Query tool. The Metadata Query tool is discussed in more detail in the next help section. In this section we will discuss accessing metadata through the datasets page.
On the datasets page, there are four links that offer up metadata in a variety of formats. The first link (html) allows you explore the heirarchical tag storm format and collapse various sections of the metadata:
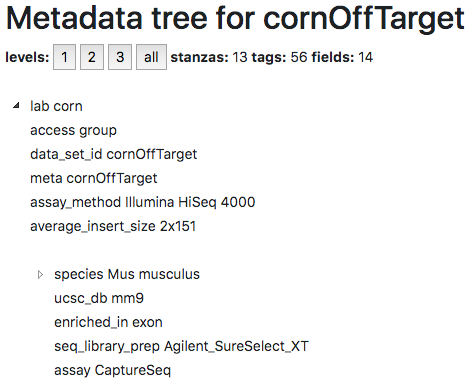
You can test this out for yourself by exploring the metadata for the kriegsteinRadialGliaStudy1 dataset.
The next three links are all to view or download the metadata in plain text formats:
Clicking these links should display the metadata in your web browser window. To download the metadata in one of these formats, you can right-click the link and select "Save Link As".
The Metadata Query tool allows to search across the metadata, quality metrics, and more for all of the datasets contained in the Stem Cell Hub. This information can be filtered and sliced using a SQL-like query interface. The output of your queries can be displayed in three different formats: (1) UCSC's heirachical tag storm (ra) format, (2) tab-separated value (tsv) format, or (3) comma-separated value (csv) format.
The interface has three boxes at the top. The first, after "select", is where you enter a comma-separated list of the metadata fields you want, e.g. lab, data_set_id, accession. Second, after "from files where" is where you put your conditionals, e.g. if you only wanted data from the Quake lab, you would enter lab="quake". Third, and last after "limit", you can enter how many records matching your query you want to display.

One example query might be pulling out some of the quality control (QC) information (enriched_in, fastq_qual_mean, fastq_qual_type, map_ratio, map_to_mouse, map_to_repeat, and map_to_ribosome fields) for the fastq files in the quakeBrainGeo1 dataset in addition to basic information such as accession, data_set_id, lab, and assay in the tsv format. This would allow us to discern the relative quality of each of these files and decide which of them meet our standards to download. The query to obtain this information would look like this:

This example will guide you through the process of finding and downloading the cell by gene expression matrix and the cell-level metadata for the single-cell dataset kriegsteinRadialGliaStudy1. It also describes some basic analysis options after you have dowloaded an expression matrix and metadata.
For this step, we will start at the File Browser and use the metadata facets to filter the visible files to display only the gene-level expression matrix for the kriegsteinRadialGliaStudy1 dataset:
As you check the boxes for these filters, you should see the files on the right-hand side of the screen slowly disappear until you see only one left. This single file should be sc002YED.tsv, the gene-level expression matrix for the kriegsteinRadialGliaStudy1 dataset.
More details about using the metadata facets for filtering can be found in the Finding data section above.
After filtering files down to just the gene-level expression matrix, sc002YED.tsv, on the File Browser for the kriegsteinRadialGliaStudy1, we will now download this file:
See the Downloading data section for information on downloading multiple files at once.
Now that you've downloaded the gene-level expression matrix, you can download the cell-level metadata for this dataset. The easiest way to this is though the Dataset Browser:
The Downloading and exploring metadata section has more information on the different ways you can download metadata.
Now that that you have a gene-level expression matrix and cell-level metadata, what next? There are many things you can do with these files from determining cellular trajectories using SCIMITAR to running dimensionality reduction and clustering algorithms in Scanpy or Seurat.
For example, you could use the UCSC Cell Browser to run dimensionality reduction and clustering algorithms from Seurat or Scanpy on your expression matrix. The results of those can then be combined with the metadata and turned into a cell browser so that the metadata and clustering can be explored interactively. The UCSC Cell Browser website provides some exemplar public datasets that you can explore.
| file | format | output | |
|---|---|---|---|
| Variant Calls | |||
| vcf | vcf | variants | |
| vcf.tbi.gz | vcf.tbi.gz | variantIndex | |
Signal data |
|||
| signal.Unique.str1.out.wig | bigWig | Plus strand unique mappings | |
| signal.Unique.str2.out.wig | bigWig | Minus strand unique mappings | |
| signal.UniqueMultiple.str1.out.wig | bigWig | Plus strand unique and multiple mappings | |
| signal.UniqueMultiple.str2.out.wig | bigWig | Minus strand unique and multiple mappings | |
| RSEM output | |||
| genes.results | text | genes | |
| isoforms.results | text | isoforms | |
STAR output |
|||
| aligned.out.bam | bam | read alignments | |
| aligned.sortedByCoord.out.bam | bam | sorted read alignments | |
| aligned.toTranscriptome.out.bam | bam | alignments of reads to transcriptome | |
| readsPerGene.out.tab | text | reads per gene | |
| SJ.out.tab | text | splicing junctions | |
| FASTQ | |||
| fastq.gz | gzipped fastq | unaligned reads |
Questions? Comments? Feel free to contact our support team.
Additional resources from the CIRM CESCG